

Počítačom podporovaný celobunkový dizajn: Integrácia syntetickej a systémovej biológie: holistický prístup
Fron. Bioeng. Biotechnol., 7. augusta 2020

Počítačom podporovaný dizajn (CAD) pre syntetickú biológiu sľubuje urýchlenie racionálneho a robustného inžinierstva biologických systémov. Vyžaduje si podrobné a kvantitatívne matematické a experimentálne modely procesov na (re)dizajn biológie, ako aj softvér a nástroje na genetické inžinierstvo a zostavovanie DNA. Zvýšená presnosť vo fáze návrhu bude mať v konečnom dôsledku dramatický vplyv na výrobu dizajnérskych buniek a organizmov s funkciami na mieru a zvýšenou modularitou. Stratégie CAD si vyžadujú kvantitatívne modely buniek, ktoré dokážu zachytiť viacrozmerné procesy a prepojiť genotypy s fenotypmi. V tomto príspevku predstavujeme pohľad na to, ako by celobunkové, viacškálové modely mohli transformovať cykly návrh – konštrukcia – test – učenie v syntetickej biológii. Ukazujeme, ako by tieto modely mohli významne pomôcť vo fázach návrhu a učenia sa a zároveň znížiť experimentálne testovanie, pričom uvádzame prípadové štúdie siahajúce od minimalizácie genómu až po bezbunkové systémy. Rozoberáme aj niekoľko výziev na realizáciu našej vízie. Možnosť opisovať a vytvárať celé bunky in silico ponúka príležitosť na vývoj čoraz automatizovanejších, presnejších a prístupnejších CAD nástrojov a stratégií.
Úvod
Celobunkové modely (WCM) sú najmodernejšími formalizmami systémovej biológie: ich cieľom je reprezentovať a integrovať všetky bunkové funkcie v jedinečnom výpočtovom rámci, čo v konečnom dôsledku umožňuje holistické a kvantitatívne pochopenie bunkovej biológie (Tomita, 2001; Karr a kol., 2015a). Kvantitatívne a vysoko výkonné experimenty in silico generované na základe WCM sľubujú výrazné skrátenie vzdialenosti medzi formuláciou hypotézy/návrhu a testovaním (Carrera a Covert, 2015).
Zatiaľ čo zjednodušené modely pre špecifické bunkové funkcie boli prvýkrát vyvinuté pred viac ako 30 rokmi [napr, regulácia expresie génov (McAdams a Arkin, 1997), signalizácia (Morton-Firth a Bray, 1998) a metabolické dráhy (Cornish-Bowden a Hofmeyr, 1991), rast buniek (Shu a Shuler, 1989) a bunkový cyklus (Goldbeter, 1991; Tyson, 1991; Novak a Tyson, 1993)], prvý WCM, model E-Cell, bol odvodený až v 90. rokoch 20. storočia pre Mycoplasma genitalium, ktorá má najmenší genóm spomedzi voľne žijúcich organizmov (Tomita a kol., 1999). Takzvaný virtuálny model samovoľne žijúcej bunky (SSC) je čiastočne stochastický; zahŕňa len podmnožinu génov kódujúcich proteíny a umožňuje dynamické simulácie, ktoré zahŕňajú rôzne subcelulárne procesy vrátane enzymatických reakcií, tvorby komplexov a translokácie látok. Súbežne s tým boli v 90. rokoch 20. storočia vyvinuté prvé metabolické modely v genómovom meradle (GSMM) Palssonovou skupinou (Varma a Palsson, 1994) s použitím analýzy bilancie tokov (FBA).
V poslednom čase boli pre rôzne organizmy zrekonštruované stovky GSMM s rastúcim počtom zastúpených génov (McCloskey et al., 2013; Yilmaz a Walhout, 2017; Mendoza et al., 2019). GSMM boli doplnené matematickým opisom ďalších procesov, ako je transkripcia, translácia a signalizácia (Lee et al., 2008; Thiele et al., 2009). Pred necelými desiatimi rokmi Covertova skupina uviedla úplnejší, hybridný WCM, ktorý reprezentuje všetky gény a molekulárne funkcie známe pre daný organizmus (Karr a kol., 2012). V tejto priekopníckej práci Karr a jeho kolegovia integrovali 28 čiastkových modelov na reprezentáciu jedného bunkového cyklu M. genitalium; každý čiastkový model je reprezentovaný osobitným formalizmom vrátane obyčajných diferenciálnych rovníc (ODE), FBA, stochastických simulácií a boolovských pravidiel.
Na zlepšenie popisnej schopnosti WCM a zvýšenie zložitosti organizmov, ktoré môžu reprezentovať, je stále potrebný značný výskum a úsilie. Vývoj WCM je náročná úloha, ktorá si vyžaduje zber rozsiahlych experimentálnych údajov, integráciu subbunkových modelov a validáciu modelov in silico/in vivo. Úplný WCM by mal v ideálnom prípade integrovať viacškálové interakcie na bunkovej úrovni (Karr a kol., 2012; King a kol., 2016) a zároveň zohľadňovať celkovú bunkovú štruktúru (Betts a Russell, 2007), dynamickú štruktúru molekulárnych interakcií (Noske a kol., 2008; McGuffee a Elcock, 2010; Yu a kol., 2016) a priestorové oddelenie subcelulárnych zložiek (Ander a kol., 2004; Takahashi a kol., 2005; Thul a kol., 2017). Zabezpečenie presného zobrazenia všetkých bunkových procesov v organizmoch s rastúcou komplexnosťou je veľmi náročné (Bouhaddou et al., 2018; Singla et al., 2018; Szigeti et al., 2018). Nie je preto prekvapujúce, že doteraz sa podarilo vytvoriť len M. genitalium a veľmi nedávno E. Coli (Macklin et al., 2020). WCM boli uvoľnené, hoci v súčasnosti sa pracuje na niekoľkých ďalších WCM1. Čitateľa odkazujeme na nedávne snahy, ktoré poskytujú prehľad o súčasnom stave vývoja WCM (Goldberg et al., 2018; Feig a Sugita, 2019).
Tu sa zameriavame na obrovský potenciál, ktorý podľa nášho názoru majú WCMs pre cykly návrhu, zostavenia a testovania integrujúce syntetickú a systémovú biológiu (obrázok 1). Hoci sú tieto aplikácie rôznorodé, majú spoločnú vysokú mieru zložitosti, ktorá by si pri absencii robustných výpočtových návrhových algoritmov založených na prediktívnych modeloch vyžadovala rozsiahle experimentálne cykly pokusov a omylov. V závere sa zamýšľame nad relevantnými výzvami, ktoré musia interdisciplinárne komunity riešiť, aby sa plne realizovala naša vízia, a diskutujeme o budúcich smeroch integrácie WCM prostredníctvom syntetickej a systémovej biológie.
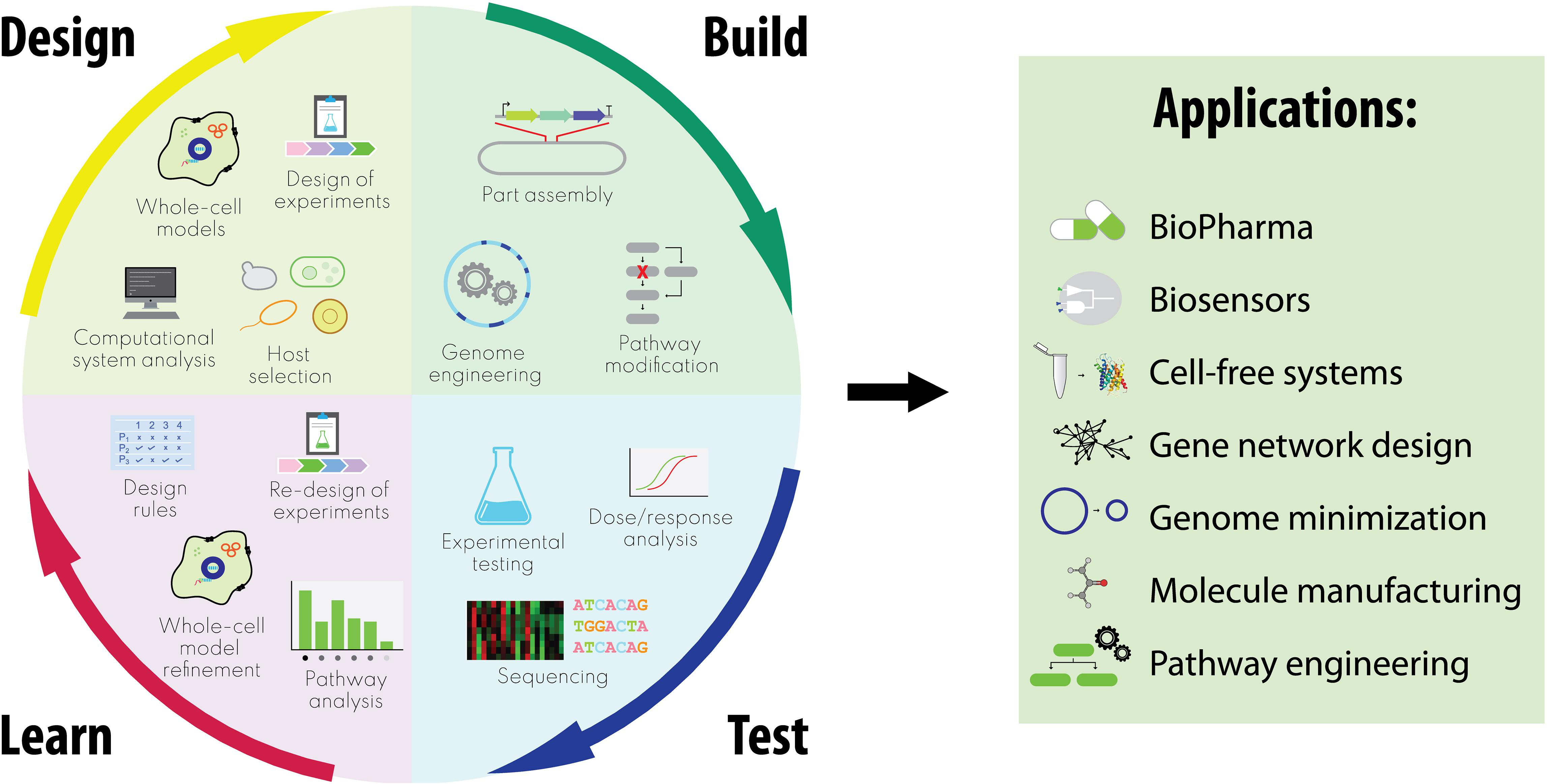
Obr. 1: Integrované cykly návrh – zostavenie – testovanie – učenie v syntetickej biológii zahŕňajúce prístupy riadené celobunkovým modelom a relatívne aplikácie.
Stratégie návrhu celých buniek v syntetickej biológii
Modelová granularita (re)návrhu génovej siete
Matematické modely môžu byť nápomocné pri (re)návrhu sieťových obvodov, ktoré rekapitulujú určité biologické funkcie. Poznatky o regulačných mechanizmoch v biologických dráhach sa získali tým, že sa živé systémy považujú za zloženie funkčných modulov, ktoré sa skúmajú prostredníctvom minimálnych počítačových modelov. Príkladom sú riadené oscilátory (Marucci et al., 2009; Purcell et al., 2010, 2013; Tomazou et al., 2018), cirkadiánne hodiny (Gerard et al., 2009; Ananthasubramaniam et al., 2020), signálne siete (Prescott a Abel, 2017), metabolizmus (Castellanos et al., 2004; Pandit et al., 2017) a transkripčná regulácia (Carrera et al., 2009). Existujúce minimálne a podrobné počítačové modely pokrývajú širokú škálu granularity biochemických detailov. Dá sa však očakávať, že ak je jadro návrhu minimálneho a podrobného modelu podobné, ich všeobecné vlastnosti sa budú zhodovať.
Pochopenie živého organizmu na úrovni systému možno dosiahnuť jeho rozložením na funkčné moduly alebo modulárne obvody (Hartwell a kol., 1999; Kitano, 2002; Ravasz a kol., 2002). Schopnosť udržať životaschopnosť prostredníctvom autonómne generovaného potomstva je nevyhnutná. Je to preto vlastnosť, ktorú WCM zohľadňujú prostredníctvom modelovania bunkového delenia, ktoré je úzko integrované s rôznymi vrstvami bunkovej regulácie (metabolizmus, signalizácia, regulácia génov, transkripcia atď.). Pre eukaryotický bunkový cyklus vypracovali Barberisova, Tysonova a Novákova skupina niekoľko minimálnych modelov (Battogtokh a Tyson, 2004; Barberis et al., 2012; Gerard et al., 2013, 2015; Linke et al., 2017; Mondeel et al., 2020).
V súčasnosti väčšine viacškálových modelov (nie WCM) chýbajú komponenty schopné premostiť bunkové siete alebo funkcie (bunkový cyklus, metabolizmus, signalizácia, regulácia génov atď.). Identifikácia uzlov, t. j. prvkov s vysokou konektivitou v bunkovom prostredí, ktoré integrujú bunkové siete, je kritickou vlastnosťou WCM. Transkripčné faktory boli nedávno identifikované ako huby, ktoré integrujú viacškálové siete, potenciálne spájajú bunkový cyklus s metabolizmom (Mondeel et al., 2019) a môžu patriť medzi časti systému, ktoré ovplyvňujú jeho stav ako celku. Vyvíjajú sa viacškálové rámce spájajúce siete rôznej granularity, a to identifikáciou relevantných regulácií vyskytujúcich sa medzi spoločnými uzlami siete a pomocou rôznych matematických formalizmov (van der Zee a Barberis, 2019). Tieto a ďalšie stratégie sa vyvíjajú aj na integráciu sietí bunkových funkčných modulov (Prescott a kol., 2015). Spolu s identifikáciou sietí, ktoré sú základom autonómnych oscilácií bunky, môžu tieto stratégie racionalizovať správne načasovanie generovania potomstva, ktoré zohľadňujú WCM.
Navrhovanie syntetických génových sietí ich modelovaním a integráciou v rámci formalizmov WCM [ako v Purcell et al. (2013)] by mohlo mať rozhodujúci význam pre skúmanie toho, ako expresia génov koreluje s používaním kodónov, skúmanie možných účinkov zaťaženia buniek (Borkowski et al., 2016) a predpovedanie modularity syntetických génových sietí a nástrojov na moduláciu expresie génov v rôznych šasi (Way et al., 2014; Pedone et al., 2019; Gomide et al., 2020).
Návrh a inžinierstvo redukovaných genómov
Minimálne genómy možno definovať ako redukované genómy obsahujúce len genetický materiál, ktorý je nevyhnutný na reprodukciu bunky (Glass et al., 2017). Štúdium a inžinierstvo minimálnych genómov môže byť nápomocné pri pochopení najdôležitejších úloh, ktoré musí bunka vykonávať na udržanie života, ako aj pri získavaní optimálnych šasi pre aplikácie syntetickej biológie s menšou záťažou buniek a vyššou odolnosťou (Moya et al., 2009; Hutchison et al., 2016; Ceroni a Ellis, 2018; Mol et al., 2018; Landon et al., 2019).
Vyčerpávajúca experimentálna charakterizácia minimalizovaného genómu je nerealizovateľná: aj v prípade takého malého organizmu, ako je M. genitalium (0,58 mb a 525 génov), existujú tisíce možných kombinácií vyradenia génov, ktoré je potrebné vykonať. Je potrebné poznamenať, že tento počet je s najväčšou pravdepodobnosťou podhodnotený, pričom sa zohľadňuje skutočnosť, že poradie, v ktorom sa vykonajú vyradenia génov, môže zmeniť výsledné fenotypy (Gawand et al., 2015). Výpočtové modely buniek v genómovom meradle by mohli byť nápomocné pri úplnom pochopení dynamickej a kontextovo závislej povahy esenciality génov (Rancati et al., 2018) a pri racionálnom navrhovaní minimalizovaných genómov in silico. Počítačové inžinierstvo minimálnych genómov by mohlo výrazne skrátiť čas a náklady na redukciu genómov v porovnaní so súčasnými prístupmi založenými na rozsiahlych experimentálnych iteráciách (Posfai et al., 2006; Iwadate et al., 2011; Hirokawa et al., 2013; Hutchison et al., 2016; Zhou et al., 2016; Reuss et al., 2017; Breuer et al., 2019).
Podľa našich najlepších vedomostí boli doteraz navrhnuté dva prístupy redukcie genómu zhora nadol založené na modeloch v genómovej mierke. Algoritmus MinGenome aplikuje algoritmus zmiešaného celočíselného lineárneho programovania (MILP) na GSMM Escherichia coli, pričom v rámci optimalizácie využíva informácie týkajúce sa esenciálnych génov a syntetických letálnych párov (Wang a Maranas, 2018). Naproti tomu Minesweeper a GAMA sú algoritmy minimalizácie genómu zhora nadol založené na WCM M. genitalium. Využívajú prístup rozdeľ a panuj, resp. skreslený genetický algoritmus na iteratívnu simuláciu redukovaných genómov (Rees-Garbutt et al., 2020); ich predpovede in silico zatiaľ neboli testované v laboratóriu.
Algoritmy redukcie genómov založené na GSMM, ako je MinGenome alebo analogické, adaptabilné metaheuristické techniky [napríklad (Burgard et al., 2003; Tang et al., 2015; Mutturi, 2017)], sú v súčasnosti širšie použiteľné v rámci organizmov vzhľadom na veľkú dostupnosť týchto formalizmov. Napriek tomu očakávame, že keď bude k dispozícii viac WCM, algoritmy na redukciu genómu založené na WCM budú poskytovať lepšie predpovede bunkových procesov a genetických interakcií vďaka bohatosti reprezentácie bunkových procesov vo viacerých mierkach.
Návrh a prototypovanie systémov bez buniek
Bezbunkové transkripčné/translačné systémy založené na surových bunkových extraktoch sú cennou platformou na riešenie základných biologických otázok kontrolovateľným a reprodukovateľným spôsobom. V posledných rokoch sa vďaka zníženiu nákladov spojených s touto technológiou a výraznému zlepšeniu možností výťažnosti syntézy (Calhoun a Swartz, 2005) stali bezbunkové systémy čoraz populárnejšie v syntetickej biológii na prototypovanie a testovanie navrhnutých biologických častí (McCloskey a kol., 2013; Reuss a kol., 2017; Yilmaz a Walhout, 2017; Mendoza a kol., 2019) a sietí (Noireaux a kol., 2003; Siegal-Gaskins a kol., 2014; Takahashi a kol., 2015). S rastúcim počtom možných aplikácií bezbunkových systémov [pozri (Silverman et al., 2020), kde je uvedený nedávny prehľad] sa vyvíjajú matematické modely na kvantitatívnu formalizáciu fungovania biologických procesov v rámci bezbunkových platforiem (Koch et al., 2018).
Doteraz boli navrhnuté deterministické modely (ODE alebo založené na obmedzeniach) na opis špecifických procesov v rámci bezbunkových platforiem, ako sú transkripcia a translácia (Karzbrun et al., 2011; Stogbauer et al., 2012; Siegal-Gaskins et al., 2014), súťaž o zdroje (Underwood et al., 2005; Borkowski et al., 2018; Matsuura et al., 2018; Moore et al., 2018) a metabolizmus (Vilkhovoy et al., 2018). Integrácia matematických formalizmov v rôznych mierkach pre bezbunkové platformy, smerujúca k WCM, by mohla byť veľmi prospešná tak na uľahčenie de novo navrhovania obvodov, ako aj na kvantitatívne porovnanie bezbunkových produktov in vitro s ich náprotivkami in vivo.
Návrh a testovanie celobunkových biosenzorov
Biosenzory sú analytické zariadenia, ktoré dokážu premeniť biochemickú reakciu na merateľný signál. Rozpoznávacia jednotka v biosenzore môže pozostávať z celých buniek, nukleových kyselín, enzýmov, proteínov, protilátok alebo ich kombinácií. Syntetická biológia výrazne urýchlila vývoj biosenzorov; bola skonštruovaná nová generácia celobunkových biosenzorov (t. j. senzorov implementovaných v celých živých bunkách), ktoré umožňujú napríklad: detekciu arzénu (Diesel a kol., 2009), detekciu znečisťujúcich látok a antibiotík (van der Meer a Belkin, 2010), detekciu mikroorganizmov v priemyselnom prostredí (Lu a kol., 2013) a diagnostické aplikácie in vivo [napr. detekciu environmentálnych signálov v črevách (Kotula a kol., 2014) a diagnostiku metastáz v pečeni (Danino a kol., 2015); prehľad pozri (Slomovic a kol., 2015)].
Aplikácia WCM na návrh, prototypovanie a testovanie celobunkových biosenzorov by mohla navrhnúť racionálne prístupy na vyladenie ich citlivosti, stability a dynamického rozsahu a zároveň uľahčiť výber ideálneho šasi a v prípade potreby usmerniť jeho prestavbu s cieľom optimalizovať výkon biosenzora (Hicks et al., 2020). Ak budú WCM dostupné pre rôzne podvozky a celé organizmy, mohli by tiež podporiť návrh optimalizovaného cieleného dodávania geneticky kódovaných biosenzorov.
Priemyselné dôsledky celobunkových modelov
Hoci intelektuálny prínos presadzovania počítačom podporovaného prístupu k celobunkovému dizajnu je nespochybniteľný, je jasné, že úspech tohto úsilia sa nakoniec bude posudzovať podľa jeho vplyvu na vedu, medicínu a priemysel. Rastúca snaha o počítačom podporované návrhy (CAD) smerom k "zeleným" chemickým prístupom, spojená s nárastom rýchlosti a schopnosti génovej syntézy a súvisiacim znižovaním nákladov, robí z biosyntézy čoraz atraktívnejšiu cestu na výrobu vysokohodnotných chemických látok (El Karoui et al., 2019). To zahŕňa množstvo príležitostí okrem iného vo farmaceutickom, agrochemickom, komoditnom chemickom a materiálovom sektore.
Veľkou výzvou však zostáva vývoj robustných, škálovateľných mikrobiálnych šasi, ktorých metabolické procesy možno predvídateľne vyladiť na požadovaný výsledok (Xu et al., 2020). V súčasnosti je výber šasi do značnej miery obmedzený na podmnožinu geneticky sledovateľných mikroorganizmov, ktorých fyziológia a výkonnosť počas fermentácie sú dobre známe a pre ktoré existujú účinné molekulárne genetické nástroje potrebné na ich manipuláciu. Optimalizácia podvozkov sa doteraz spoliehala výlučne na postupné, postupné zlepšovanie požadovaných vlastností hostiteľského kmeňa vrátane rýchlosti rastu, využitia vstupných surovín a výťažku produktu (Calero a Nikel, 2019). Z týchto dôvodov zostáva proces optimalizácie šasi neúnosne pomalý a drahý, čo sčasti spôsobuje nedostatok vysokohodnotných malých molekúl, ktoré sa v súčasnosti vyrábajú pomocou procesov syntetickej biológie. Cielené manipulácie často vedú k neočakávaným necieľovým účinkom, ktoré súvisia so spoluzávislosťou metabolických procesov, ktoré vo všeobecnosti fungujú v súčinnosti v rámci vzájomne závislých bunkových sietí (Woolston a kol., 2013): narušenia môžu skôr ohroziť ako zlepšiť želané vlastnosti, čo vedie k neželaným výsledkom. Je zrejmé, že robustné, predvídateľné WCM predstavujú atraktívne riešenie problému optimalizácie podvozkov, pretože poskytujú univerzálny nástroj, ktorý možno použiť na odstránenie závislostí a zabezpečenie splnenia kritérií výkonnosti.
Okrem toho je potrebné vyriešiť zložitosti spojené s heterogenitou populácie počas fermentácie podvozkov (Danchin, 2012). Aby boli priemyselné procesy založené na fermentácii uskutočniteľné, musia byť výťažky produktov dostatočne vysoké, aby bola biosyntéza finančne rentabilná. Vznik "podvodníkov" alebo pomaly rastúcich mikroorganizmov v rámci mikrobiálnych populácií by sa mal riešiť pomocou nastaviteľných regulačných procesov, ktoré fungujú v rámci celej populácie. Zavedenie takýchto vlastností je hlavnou výzvou pre konvenčné prístupy k návrhu podvozkov. Prístupy založené na WCM by mohli ľahšie zaviesť a testovať tieto procesy.
Rozhodujúca pre úspech počítačom podporovaného prístupu návrhu celých buniek je kvalita použitého modelu (Fernandez-Castane et al., 2014). Mikrobiálne systémy s malými genómami predstavujú presvedčivý vstupný bod pre štúdium, pričom vývoj modelu by mohli uľahčiť prebiehajúce štúdie zamerané na stanovenie základných zložiek funkčného genómu. Tieto štúdie sú sčasti poháňané experimentmi s minimalizáciou genómu, ktoré sa zase môžu použiť na ďalšie zdokonaľovanie výkonnosti modelu. Dôležité je, že v našom chápaní mikrobiálnych metabolických procesov pretrvávajú zásadné medzery, čo bude nepochybne brániť pokroku (Price et al., 2018). Schopnosť WCM predpovedať predtým neidentifikované metabolické závislosti by sa však mala považovať za kyslý test platnosti modelu. GSMM totiž často zlyhávajú kvôli neschopnosti zohľadniť metabolické závislosti, čo viedlo k skepse v priemyselných kruhoch, ktoré spochybňujú hodnotu takýchto modelov. Celobunkové prístupy ponúkajú mechanizmus na obídenie tohto problému. To má osobitný význam pri vývoji šasi pre "neprirodzené" produkty, ktorých chemizmus sa vymyká metabolitom, ktoré sa nachádzajú v prírode (Calero a Nikel, 2019). Rozširovanie metabolickej kapacity organizmov na šasi s cieľom dodávať takéto nové produkty predstavuje riziko zavedenia ďalších zložitostí vrátane nadmerného vyčerpania základných zásobníkov metabolitov alebo tvorby toxických produktov alebo medziproduktov. Konštrukčné prístupy riadené WCM majú jedinečnú možnosť identifikovať takéto problémy a poskytnúť cestu k ich obchádzaniu.
Schopnosť navrhnúť explicitnú kontrolu nad správaním buniek je tiež rozhodujúca pre priemyselné prijatie modelových podvozkov. Možno tvrdiť, že schopnosť regulovať bunkové procesy je rovnako dôležitá ako definovanie samotných procesov. Nastaviteľné regulačné systémy musia umožňovať určitý stupeň vnútornej aj vonkajšej kontroly. Prístupy založené na syntetickej biológii na konštrukciu genetických obvodov nás v súčasnosti stavajú na cestu k širokospektrálnej bunkovej regulácii, hoci stále existujú problémy. Tieto systémy sú často nedostatočne ortogonálne, pričom pre rôzne šasi sú potrebné individuálne návrhy v dôsledku rozdielov v základnom metabolickom procese (Pandit a kol., 2017). Riešenie tohto problému opäť ponúkajú prístupy celobunkového dizajnu, keďže takéto systémy možno preddefinovať a otestovať ich funkčnosť in silico pred uskutočnením nákladných laboratórnych experimentov.
Čo ďalej? Prekročenie hranice prototypu
V posledných rokoch pokrok v technológiách genomických meraní na generovanie údajov, vytvorenie dátových úložísk a vývoj simulačných platforiem WCM výrazne uľahčili odvodenie WCM [pozri prehľad (Goldberg et al., 2018)]. Napriek tomu si implementácia cyklov WCM založených na navrhovaní, zostavovaní a testovaní pre inžinierstvo v genómovom meradle vyžaduje riešenie ďalších výziev [Bartley et al., 2020].
Ak sa má model použiť na návrh a prototypovanie inžinierskeho živého systému, musí byť model spoľahlivý. Dokonca aj v prípade jednoduchého organizmu sa počet kinetických parametrov zvyšuje so zvyšujúcou sa zložitosťou a úrovňou podrobnosti matematického modelu; obmedzenie parametrov sa tak stáva ťažším a vyžaduje si rozsiahle experimentálne údaje. Matematické modely možno použiť na vytvorenie predpovedí chýbajúcich údajov, často však abstrahujú fyzikálne procesy pomocou zjednodušujúcich predpokladov, ktoré môžu platiť v špecifických podmienkach (Babtie a Stumpf, 2017). Na stanovenie 1 462 kvantitatívnych parametrov WCM M. genitalium boli z dôvodu nedostatku údajov špecifických pre organizmus zahrnuté hodnoty z príbuzných organizmov (Macklin a kol., 2014); vykonala sa kombinácia hodnôt parametrov uvedených z predchádzajúcich experimentov a numerická optimalizácia na redukovanom modeli. Hoci by sme v ideálnom prípade chceli merať všetky kinetické parametre priamo z experimentov, stále nám chýba možnosť merať každý stav v jednotlivých bunkách v priebehu času a vo všetkých možných podmienkach prostredia. Pre formalizmy v genómovom meradle a WCM bude pravdepodobne potrebná kombinácia priameho experimentálneho odhadu a odvodenia parametrov.
Analýza citlivosti, ktorá sa zvyčajne vykonáva perturbovaním parametrov s cieľom pochopiť, ako neistoty ovplyvňujú výstupy modelu (Erguler a Stumpf, 2011), môže byť pri aplikácii na modely v genómovom meradle extrémne výpočtovo náročná. Alternatívou by mohli byť štatistické prístupy, ako napríklad prístupy založené na Bayesovských metódach (Vernon et al., 2018) alebo Fisherovej informačnej matici (Rand, 2008), ktoré by sa mohli opatrne vykonávať aspoň na úrovni čiastkových modelov a prípadne rozšíriť na WCM. Na vývoj nových techník odhadu parametrov špecifických pre WCM bola zorganizovaná výzva Reverse Engineering Assessments and Methods (DREAM8) (Karr et al., 2015b). Navrhla možné zaujímavé cesty pre parametrizáciu WCM (t. j. redukciu modelu a kombináciu diferenciálnej evolúcie a náhodných lesov) a zdôraznila, že dostupnosť komplexných údajov je rozhodujúca na zabezpečenie praktickej identifikovateľnosti modelu (Ashyraliyev et al., 2009) a na kalibráciu WCM.
Výskumníci začali zhromažďovať údaje potrebné na vývoj WCM do verejných repozitárov [napríklad (Wittig et al., 2012; Kolesnikov et al., 2015; Sajed et al., 2016; UniProt Consortium, 2018; Caspi et al., 2020)]; napriek tomu sú údaje potrebné na odvodenie a prispôsobenie WCM rozptýlené v mnohých repozitároch a publikáciách a často nie sú anotované alebo normalizované, čo si v konečnom dôsledku vyžaduje obrovské manuálne úsilie. Existujú aj federatívne archívy repozitárov, ako je napríklad systém PDB-Dev na ukladanie integračných/hybridných modelov a zodpovedajúcich údajov (Burley et al., 2017), ktoré by mohli byť vhodné na archiváciu a šírenie údajov aj modelov a zároveň by umožnili rôznym výskumníkom pokúšať sa o alternatívne prístupy k modelovaniu/parametrizácii. Covertova skupina vyvinula databázu WholeCellKB (Karr et al., 2013) na organizáciu kvantitatívnych meraní (viac ako 1 400), z ktorých bol odvodený WCM M. genitalium; bolo by ideálne umožniť automatický prístup a vyhľadávanie v takýchto databázach.
Na zlepšenie reprodukovateľnosti WCM a spolupráce sú potrebné aj nové normy a simulačný softvér (Medley et al., 2016). Výskumníci by mali investovať úsilie do využívania a rozširovania možností štandardných formátov, ako je napríklad Systems Biology Markup Language (SBML) (Hucka et al., 2003) a Systems Biology Graphical Notation (SBGN) (Le Novere et al., 2009), aby boli vhodné pre WCM. Napríklad viaceré aspekty WCM M. genitalium nie je možné reprezentovať pomocou SBML, ako napríklad multialgoritmický charakter modelu (Waltemath et al., 2016). Na umožnenie reprodukovateľných simulácií WCM je potrebný ďalší vývoj štandardných formátov modelovania, napr. zahrnutím ontológií do balíka SMBL Hierarchical Model Composition, ktoré by mohli reprezentovať algoritmus potrebný pre konkrétne čiastkové modely (Courtot et al., 2011). V kontexte aplikácií syntetickej biológie sa domnievame, že by bolo vhodné a prospešné uvádzať a ukladať údaje týkajúce sa rôznych iterácií predpovedí vytvorených WCM in silico, testovania in vivo a prípadného zdokonaľovania modelu/návrhu; tým by sa stanovila prediktívna sila WCM a osvetlili by sa kroky na zefektívnenie cyklov návrh - zostavenie - testovanie - učenie.
Dôležité je tiež zvážiť štrukturálne neistoty modelu, ktoré závisia od predpokladov modelu. Zatiaľ čo pre určité súbory modelov (napr. malé ODE systémy pre signálne dráhy) boli na výber modelov navrhnuté prístupy založené na pravdepodobnosti a bayesovskom prístupe (Wilkinson, 2007; Kirk et al., 2013) a semidefinitné programovanie na zneplatnenie modelu (Anderson a Papachristodoulou, 2009), pre WCM neboli doteraz navrhnuté žiadne vhodné techniky.
Predpokladáme, že automatizácia bude zohrávať zásadnú úlohu pri odvodzovaní WCM pre eukaryotické organizmy a pri ich aplikácii na navrhovanie komplexných procesov. V ideálnom prípade by sme chceli zaviesť automatizáciu v rôznych fázach, ako je extrakcia údajov z literatúry, odvodenie modelu a integrácia modelu/údajov v rámci krokov prispôsobenia modelu a validácie, ako aj pri porovnávaní predpovedí dizajnu in silico s testami in vivo (Bartley et al., 2020). To si zasa bude vyžadovať prijatie štandardov pre úložiská údajov aj modelov. Očakáva sa tiež, že automatizácia laboratórií v spojení s CAD založeným na WCM zmení cykly návrhu, tvorby a testovania. Keďže používanie robotiky sa stáva čoraz bežnejším na akademickej pôde aj v priemysle, môže sa výrazne zvýšiť priepustnosť a reprodukovateľnosť experimentov potrebných na odvodenie aj validáciu WCM a uľahčiť zdieľanie protokolov medzi výskumnými komunitami (Jessop-Fabre a Sonnenschein, 2019).
Na pomoc pri zavádzaní WCM pre aplikácie v syntetickej biológii sú potrebné vysoko výkonné paralelizované počítačové klastre na spúšťanie modelov s dlhým časom behu, koordináciu príslušných databáz, parametrizáciu a validáciu modelov a následné začlenenie WCM do návrhových cyklov v kombinácii s optimalizačnými algoritmami (Macklin a kol., 2014; Chalkley a kol., 2019).
Zavedenie štandardizovaných nástrojov na zdieľanie údajov a simuláciu WCM by zase uľahčilo validáciu modelov. To by malo zahŕňať definovanie vhodných metrík a techník formálnej verifikácie modelov, ako sú techniky vyvinuté pre modely kódované v jazyku SBML (Kwiatkowska et al., 2011).
(Pre)myslenie systémových prístupov: Spoločné úsilie
Pri riešení uvedených výziev sa domnievame, že existuje obrovská príležitosť prehodnotiť doteraz používané prístupy na vytváranie modelov v genómovom meradle vrátane WCM a integrovať sa so širšími komunitami vrátane softvérových inžinierov, informatikov, štrukturálnych biológov, bioinformatikov a systémových a syntetických biológov.
Predpokladáme, že pri synergii rôznych komunít v oblasti výskumu súvisiaceho s WCM by sa do modelov v genómovom meradle mohli integrovať rôzne druhy formalizmov. Symbolické uvažovanie poskytuje celý rad expresívnych a intuitívnych logických rámcov, ktoré by potenciálne mohli dopĺňať a pomáhať spájať čiastkové modely v rôznych mierkach. Takéto metódy sa bežne používajú na komplexné systémy v elektronickom a softvérovom priemysle a už takmer desať rokov sa aplikujú na biologické systémy (Iyengar, 2011). Nedávne práce ukázali možnosť uplatnenia metód logického programovania na signálne dráhy (Ray a kol., 2011), metabolické siete (Bragagli a Ray, 2015) a automatizáciu mechanistickej filozofie vedeckého objavu v simulovaných organizmoch (Rozanski a kol., 2015); malo by byť možné integrovať takéto čiastkové modely v rámci WCM.
Domnievame sa, že existuje priestor na ďalšie zvýšenie deskriptívnej a prediktívnej schopnosti WCM v priestorových a časových mierkach integráciou komunít štrukturálnej biológie a molekulárneho modelovania s cieľom dôkladne zohľadniť nielen biochemické, ale aj fyzikálne, molekulárne a štrukturálne zložky buniek. Vývoj takzvaných "fyzikálnych" WCM [pozri (Feig a Sugita, 2019) a (Feig a Sugita, 2013) pre komplexné prehľady] je rozvíjajúcou sa oblasťou, pričom prvé modely opisujú minimálne bunkové prostredia v úplných atomistických detailoch (Feig a kol., 2015; Yu a kol., 2016). S konečným cieľom integrovať biochemické a fyzikálne WCM v rámci multiškálového rámca (Sali et al., 2015) potrebujeme prístupy, ktoré sa dokážu vyrovnať s obmedzeniami atomistických modelov biomolekúl (najmä z hľadiska výpočtových zdrojov), prípadne využiť hrubozrnné (Ando a Skolnick, 2010; Hyeon a Thirumalai, 2011) alebo kontinuálne (Solernou et al., 2018) prístupy.
V spolupráci so softvérovými inžiniermi je potrebné vyvinúť nástroje, ktoré umožnia a prípadne zautomatizujú integráciu rôznych typov údajov v rôznych mierkach, odvodenie, prispôsobenie a overenie modelov a vizualizáciu a interpretáciu výsledkov (Szigeti a kol., 2018).
Okrem toho by sa modely založené na pravidlách mohli stať novým štandardom na reprezentáciu každého molekulárneho druhu s požadovanou úrovňou granularity a multialgoritmických čiastkových modelov (napr. FBA a stochastické dynamické modely). Nedávno sa začali vyvíjať rámce, v ktorých je intuitívna logika spojená s modelmi založenými na pravidlách (van der Zee a Barberis, 2019).
Keďže produkujeme čoraz väčšie množstvo experimentálnych údajov a čoraz sofistikovanejšie výpočtové nástroje na realizáciu podrobných a komplexných reprezentácií skutočných buniek, prístupy zamerané namiesto toho na zámerne abstraktné a úsporné simulácie umelých bunkových systémov poskytujú cennú zmenu perspektívy. Takéto "hračkárske modely" by mohli byť cenným nástrojom na testovanie rôznych algoritmov na odvodenie a prispôsobenie modelov a zároveň by mohli ponúknuť príležitosť na zapojenie širších výskumných komunít a verejnosti (Castiglione a kol., 2014).
Napokon sa domnievame, že existuje obrovský potenciál na uplatnenie techník strojového učenia pri odvodzovaní WCM, ako aj pri ich aplikáciách v syntetickej biológii. Dve nedávne práce (Lin et al., 2017; Ma et al., 2018) ukázali, že hlboké neurónové siete majú dobré predpoklady na rekonštrukciu architektúry živých systémov [konkrétne hierarchickej organizácie jadrových transkripčných faktorov v jadre (Lin et al., 2017) a základnej eukaryotickej bunky (Ma et al., 2018)] a predpovedanie bunkových stavov a fenotypov. V oboch prípadoch bola konfigurácia vrstiev siete, a teda aj biologická štruktúra, formulovaná s využitím rozsiahlych predchádzajúcich znalostí, čo v konečnom dôsledku umožnilo plne "viditeľné" systémy, v ktorých možno mechanisticky skúmať všetky vnútorné biologické stavy (Yu et al., 2018). Strojové učenie by mohlo byť prospešné pri systematickom spracovaní veľkých súborov celobunkových údajov in vivo a in silico, napríklad použitím bayesovskej inferencie, na integráciu údajov z rôznych zdrojov a doplnenie riedkych údajov (Perdikaris a Karniadakis, 2016) a na pomoc pri automatickej klasifikácii simulácií WCM a prepojení fenotypov s genotypmi (Alber et al., 2019). Metódy ansámblov, ktoré spájajú viacero nezávislých modelov do jedného prediktívneho modelu na zvýšenie celkovej robustnosti predpovedí, by sa mohli prijať aj na vývoj subcelulárnych formalizmov a podporu ich integrácie v rámci šasi (Camacho et al., 2018). Okrem toho by pri identifikácii parametrov WCM mohlo pomôcť strojové učenie, napríklad použitím bayesovského odhadu parametrov (Vyshemirsky a Girolami, 2008), regresných modelov a techník posilneného učenia (Alber et al., 2019). Cennú metodiku na výber najlepších experimentálnych súborov údajov na identifikáciu aj validáciu modelu by mohli ponúknuť aj techniky optimálneho experimentálneho dizajnu (Smucker et al., 2018).
Diskusia
Ukázali sme, že WCM budú pravdepodobne nápomocné pri informovaní o cykloch návrhu, tvorby a testovania v aplikáciách syntetickej biológie. WCM môžu urýchliť realizáciu "dizajnérskych" buniek a organizmov prispôsobených špecifickým funkciám, znížiť počet experimentálnych iterácií a zvýšiť predikčnú silu doteraz používaných výpočtových formalizmov.
Pri (re)dizajne funkcií bunkových sietí je preto dôležité kvantitatívne analyzovať a predpovedať prostredníctvom špecializovaných modelovacích stratégií dynamiku interakcií medzi rôznymi vrstvami bunkovej regulácie. WCM by teda mali zohľadňovať, ako sú rôzne bunkové vrstvy integrované a ako prebieha regulačná spätná väzba medzi týmito vrstvami v čase. Tieto výzvy sa musia riešiť prostredníctvom integračného výpočtového a experimentálneho spoločného úsilia zameraného na: (i) inžinierske návrhy sietí in vivo, ktoré prostredníctvom prediktívnej systémovej biológie môžu byť schopné autonómne oscilovať a udržiavať generáciu potomstva, a (ii) extrakciu, vizualizáciu a funkčný prieskum regulačných interakcií medzi bunkovými vrstvami prostredníctvom nových viacškálových modelov.
Keďže syntetická biológia smeruje k (re)inžinierstvu celých genómov a viacbunkových systémov, interdisciplinárne komunity musia spolupracovať na vývoji nástrojov, ktoré sú potrebné na zlepšenie prediktívnej sily WCM. Hoci výzvy pretrvávajú, je zrejmé, že prijatie metód založených na modeloch má potenciál zmeniť základný výskum aj súčasný proces vývoja bioprodukcie, čo povedie k výraznému zlepšeniu výkonnosti hostiteľa a výťažnosti produktov v priemyselnom meradle.
V konečnom dôsledku, keď sa vývoj kinetických modelov v rozsahu ľudského genómu stane uskutočniteľnejším (Bordbar et al., 2015; Szigeti et al., 2018), predpokladáme, že celobunkové formalizmy sa stanú nepostrádateľným nástrojom na štúdium ľudských variácií a navrhovanie liečebných postupov a syntetických bunkových skríningových systémov.